URGENT MEDICAL DEVICE CORRECTION

-
PENTAX Medical endoscopic reprocessing instruction for use (rIFU)
Dear Healthcare Professional,
This letter is to inform you that PENTAX Medical (“PENTAX”) is conducting a voluntary corrective action of impacted PENTAX Medical Video Upper GI Scopes (EG) and Video Colonoscopes (EC) families related to reprocessing endoscopic units.
These endoscopic units are Class II devices and offered for sale in the USA. PENTAX Medical has identified issues with the reprocessing IFUs (rIFUs) where the reprocessing user would have to identify what configuration endoscope is being worked on and to ensure the reprocessing is performed in a safe and effective manner. As a result, PENTAX separated the EC and EG rIFUs into 5 separate rIFUs and conducted a Human Factor study and subsequently cleared by the FDA. This regrouping of rIFUs did not require changes to the reprocessing instructions for use, nor were there any changes to the design, intended use, or indications for use of the EG family and EC family of endoscopes.
The following models are included in this corrective action:
Colonoscope Family Description Endoscope Family Description Colonoscope Models Colonoscope Family # 1 Colonoscopes with One Instrument Channel and a Water Jet Channel EC-2990Li, EC-3490Li, EC-3890Li, EC-3490TLi, EC-3490LK, EC-3890LK, EC3890LZi, EC34-i10L, EC38-i10L Colonoscope Family # 2 Colonoscopes with Two Instrument Channels and a Water Jet Channel EC-3890TLK Gastroscope Family Description Endoscope Family Description Gastroscope Models Gastroscope Family # 1 Gastroscopes without a Water Jet Channel EG-2790i, EG-1690K, EG-2490K, EG-2790K, EG27-i10 Gastroscope Family # 2 Gastroscopes with a Water Jet Channel EG-2990i, EG-2990K, EG-3490K, EG29-i10 Gastroscope Family # 3 Gastroscopes with Two Instrument Channels and a Water Jet Channel EG-3890TK The grouping will allow the following for reprocessing personnel:
- Eliminate the need to identify optional configurations of instrument channels and water jet channel
- Simplify the instructions and graphics in the individual rIFUs
Please replace any previous versions of rIFUs with the most current revision. Please note that this documentation set is current as of this date. For future updates, please refer to the PENTAX online IFU library at https://ifu.pentaxmedical.com.
Customer Instructions: Please complete the enclosed Field Correction Response Form upon receipt of this package, and email to PENTAX at customeradvisories@pentaxmedical.com.
Adverse events experienced with the use of this product should be reported as soon as possible to PENTAX at vigilance@pentaxmedical.com.
Contact Information: PENTAX regrets any inconvenience that this action may cause and appreciates your understanding and cooperation. PENTAX will issue additional communications as further information becomes available. Please be assured that maintaining patient safety and quality is our utmost priority.
If you have any questions regarding this action, please feel free to contact us at:- Tel: 1-800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST)
- Fax: 201-799-4063 (alternate 201-391-4189)
- Email: customeradvisories@pentaxmedical.com
Sincerely
PENTAX Medical
FIELD CORRECTION RESPONSE FORM
-
URGENT MEDICAL DEVICE CORRECTION - PENTAX Medical ED34-i10T2 duodenoscope instructions for use (IFU)
Dear Healthcare Professional,
This letter is to inform you that PENTAX Medical (“PENTAX”) is conducting a voluntary corrective action of PENTAX Medical ED34-i10T2 duodenoscope units.
These duodenoscope units are Class II devices and offered for sale in the USA. PENTAX Medical has identified an issue in relation to the application of the OE-A63 (Single-Use Sterile Distal End Cap with Elevator from the ED34-i10T2 duodenoscope). If not properly attached and verified per the instructions for use, this end cap can unexpectedly fall off of the duodenoscope during procedures. This can result in unforseen events such as mucosal injury, lacerations, or bleeding of the patient. Detachment of the distal end cap (OE-A63) into the oral cavity of the patient may also result in aspiration.
As a result, we have updated our instructions for use (IFU) for both the OE-A63 distal end cap and ED34-i10T2 duodenoscope. The warning section of the IFU has been updated to notify users of the associated risks with the distal end cap (OE-A63) unexpectedly becoming detached during a procedure. In addition, the IFU has been updated to notify users of what immediate actions should be taken in case the event occurs.
Please note that this documentation set is current as of this date. For updates, please refer to the PENTAX online IFU library at https://ifu.pentaxmedical.com.
Customer Instructions: Please complete the enclosed Field Correction Response Form upon receipt of this package, and email to PENTAX at customeradvisories@pentaxmedical.com.
Adverse events experienced with the use of this product should be reported as soon as possible to PENTAX at vigilance@pentaxmedical.com.
Contact Information: PENTAX regrets any inconvenience that this action may cause and appreciates your understanding and cooperation. PENTAX will issue additional communications as further information becomes available. Please be assured that maintaining patient safety and quality is our utmost priority.
If you have any questions regarding this action, please feel free to contact us at:
- Tel: 1-800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST)
- Fax: 201-799-4063 (alternate 201-391-4189)
- Email: customeradvisories@pentaxmedical.com
FIELD CORRECTION RESPONSE FORM
-
URGENT MEDICAL DEVICE CORRECTION - PENTAX Medical “FDA-cleared” endoscopic instructions for use (IFU) and reprocessing instruction for use (RIFU)
Dear Healthcare Professional,
This letter is to inform you that PENTAX Medical (“PENTAX”) is conducting a voluntary corrective action of impacted endoscopic units.
These endoscopic units are Class I exempt and/or Class II devices and offered for sale in the USA. As part of a correction to a FDA regulatory compliance action PENTAX observed certain gaps in record keeping. As a result, we are unable to verify that in some cases, our customers received the latest version of operating instruction for use/reprocessing instruction for use (OIFU/RIFU) documentation. PENTAX is sharing the current OIFU/RIFU with customers in order to assure that the most current versions of these documents are in use. This is being provided electronically on the USB drive along with the enclosed instruction sheet.
Please note that this documentation set is current as of this date. For updates, please refer to the PENTAX online IFU library at https://ifu.pentaxmedical.com.
Customer Instructions:Please complete the enclosed Field Correction Response Form upon receipt of this package, and email to PENTAX at customeradvisories@pentaxmedical.com.
Adverse events experienced with the use of this product should be reported as soon as possible to PENTAX at vigilance@pentaxmedical.com.
Contact Information: PENTAX regrets any inconvenience that this action may cause and appreciates your understanding and cooperation. PENTAX will issue additional communications as further information becomes available. Please be assured that maintaining patient safety and quality is our utmost priority.
If you have any questions regarding this action, please feel free to contact us at:
o Tel: 1-800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST)
o Fax: 201-799-4063 (alternate 201-391-4189)
o Email: customeradvisories@pentaxmedical.com
Sincerely,
PENTAX Medical
Supplementary Documents:
-
URGENT MEDICAL DEVICE CORRECTION - PENTAX Medical 9372HD Digital Capture version 1.1.0 or higher and 9310HD Digital Video Capture Module with software version 3.4.0 or higher, both configured with 9263 endoPortal™
October 22, 2020
Dear Healthcare Professional,
This letter is a clarification of and replaces the letter dated September 2, 2020, which you received previously.
This letter is to inform you that PENTAX Medical (“PENTAX”) is conducting a voluntary corrective action of impacted 9372HD Digital Capture version 1.1.0 or higher and 9310HD Digital Video Capture Module with software version 3.4.0 or higher, both configured with 9263 endoPortal™.
9372HD Digital Capture and 9310HD Digital Video Capture Module are Class I exempt devices and offered for sale in the USA.
PENTAX Medical has identified an intermittent software issue that could affect your system, in which an exam video for one patient (Patient A) might be copied to another patient (Patient B). This does not occur at the time you perform and initially review an exam; it is not evident until a follow-up review occurs.
As a consequence, at a later date:
- You cannot review the video for Patient A.
- When you review a video for Patient B, the system plays the incorrect file. The audio portion might provide an indication that the file is incorrect.
This issue does not happen during the live exam and the initial patient evaluation. The problem is noticed intermittently when reviewing an exam that has been moved to the archived video location. During the follow-up exam, the assessment of the patient's progress could be affected when comparing videos.
Note: the still images captured and patients reports performed at the initial time of the exam will have the correct images.
Customer Instructions: Please complete the enclosed Field Correction Response Form which identifies the serial numbers of the affected products your facility owns.
PENTAX Medical will be taking a two-step approach to contain and correct the problem.
- First, we will contact you to schedule a time for our Field Service to temporarily disable file transfer to endoPortal.
- During the time that file transfers to endoPortal are disabled, you can continue to create and save exam video files. When this issue is resolved, all files recorded during that period will be uploaded and will be available for endoPortal.
- Second, PENTAX Medical is developing a software solution that will be deployed remotely. We will coordinate all updates with your IT administrator.
- Adverse events experienced with the use of this product should be reported as soon as possible to PENTAX at vigilance@pentaxmedical.com.
- Contact Information: PENTAX regrets any inconvenience that this action may cause and appreciates your understanding and cooperation. PENTAX will issue additional communications as further information becomes available. Please be assured that maintaining patient safety and quality is our utmost priority.
If you have any questions regarding this Action, please feel free to contact us at: Tel: 1-800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST) Fax: 201-799-4063 (alternate 201-391-4189) Email: customeradvisories@pentaxmedical.com.
Sincerely,
PENTAX Medical
Supplementary Documents:
-
URGENT MEDICAL DEVICE CORRECTION - Andorate® Disposable Accessory Valves Set GAR081P, GAR037P & GAR037C For PENTAX 90/i10/i10c Series Endoscopes
Dear Healthcare Professional,
This letter is to inform you that PENTAX Medical (“PENTAX”) is conducting a voluntary corrective action of impacted Andorate® Disposable Accessory Valves Set GAR081P, GAR037P & GAR037C For PENTAX 90/i10/i10c Series Endoscopes.
Andorate® Disposable Accessory Valves Set GAR081P, GAR037P & GAR037C for PENTAX 90 Series Endoscopes are Class II devices (Accessories to endoscopes) and offered for sale in the USA. PENTAX Medical is a distributor of the Andorate® Disposable Accessory Valve Set, manufactured by GA Health Company Limited, for use with Pentax 90/i10/i10c series endoscopes. The disposable Andorate Valve Set (Model GAR081P) consists of disposable Andorate Valve Set (Model GAR037) and the Andorate Auxiliary Water Connector (Model GAR048) that are packaged together. The Andorate® Valve Set is intended for single-use and is supplied sterile.
PENTAX Medical was made aware of multiple complaints about a defective Andorate Disposable Suction Control Valve that is a component of the Andorate® Valve Set and is designed to control the suction function of an endoscope during a GI endoscopic procedure. The problem may occur with the suction valve after it is depressed and does not return to its original position. Valves are depressed by doctors and not returning to a full closed position without the physician pulling back up on the valve, which results in continuous suction.
The root cause of this issue was identified as compromised performance of the Andorate disposable suction valves, and not due to any malfunction of the endoscopes. No serious injury or death of a patient or user, which would require medical intervention was reported. However, this issue can lead to, or has the potential to lead to, temporary discomfort or product complaints or some other form of dissatisfaction.
Customer Instructions: Enclosed with this letter is a Field Correction Response Form which identifies the Product and Quantity of the affected products your facility owns.
PENTAX Medical will be taking the following approach to contain and correct the problem.
The affected product GAR081P, GAR037P & GAR037C Andorate® Disposable Suction Valves Set will be removed from the market.
Model
Affected Lot Number
GAR081P
19110102, 19121608,19121623
GAR037C
19120907
GAR037P
19110101
Adverse events experienced with the use of this product should be reported as soon as possible to PENTAX at vigilance@pentaxmedical.com.
Contact Information: PENTAX regrets any inconvenience that this action may cause and appreciates your understanding and cooperation. PENTAX will issue additional communications as further information becomes available. Please be assured that maintaining patient safety and quality is our utmost priority.
If you have any questions regarding this action, please feel free to contact us at: Tel: 1-800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST) Fax: 201-799-4063 (alternate 201-391-4189) Email: customeradvisories@pentaxmedical.com
Sincerely,
PENTAX Medical
Supplementary Documents:
Field Correction Response Form
-
URGENT MEDICAL DEVICE CORRECTION AND REMOVAL - PENTAX Medical Colonoscopes model EC34-i10L and EC38-i10L
Re: PENTAX Medical Colonoscopes model EC34-i10L and EC38-i10L
Dear Healthcare Professional,
This letter is to inform you that PENTAX Medical (“PENTAX”) is conducting a voluntary recall/corrective action of all EC34-i10L and EC38-i10L Colonoscopes with serial number beging with the letter L due to a stock mixup.
EC34-i10L & EC38-i10L were cleared by FDA on K131855 and offered for sale in the USA.
The Japan Design group made changes to the unit designs for units with serial numbers beginning with the letter L;
- EC34-i10L the design of the internal lumen of the Water Nozzle was changed to improve cleaning of the objective lens.
- EC38-I10L the design of the internal lumen of the Water Nozzle was changed to improve cleaning of the objective lens and the positions of the Air and Water Nozzles with respect to the Objective Lens were reversed.
The impact of the design changes were evaluated by the design group and thought to be not significant to reprocessing efficacy. There were no changes required to the reprocessing instructions texts for the units as the changes were to the internal design of the unit. Pentax of America Inc Regulatory Group assessed the changes based on FDA guidance and determined that a 510(k) should be submitted for the design change. Five units of the changed design were distributed in the USA in error.
Customer Instructions: Enclosed with this letter is a Field Correction Response Form which identifies the serial numbers of the affected products your facility owns. Please quarantine the units your facility owns, complete the response form, and return it to PENTAX Medical using the e-mail address or fax number listed below.
Upon return of the response form, PENTAX will contact your facility to arrange the return of the affected units. Loaner units will be supplied until this issue is resolved. .
Adverse events experienced with the use of this product should be reported as soon as possible to PENTAX at vigilance@pentaxmedical.com and the FDA’s MedWatch Adverse Event Reporting program.
Contact Information: PENTAX regrets any inconvenience that this action may cause and appreciates your understanding and cooperation. PENTAX will issue additional communications as further information becomes available. Please be assured that maintaining patient safety and quality is our utmost priority.
If you have any questions regarding this action, please feel free to contact us at: Tel: 1-800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST) Fax: 201-799-4063 (alternate 201-391-4189) Email: customeradvisories@pentaxmedical.com
Sincerely,
PENTAX Medical
AFFECTED UNITS
SERIAL NUMBER
EC34-I10L
L110300
EC34-I10L
L110306
EC34-I10L
L110301
EC34-I10L
L110303
EC38-I10L
L110746
-
Customer Information on Petya Ransomware for PENTAX Medical Informatics, Speech, Voice and Swallowing Systems
Description
We recognize our Informatics, Speech, Voice and Swallowing Systems are impacted by the Petya ransomware virus. Petya is designed to spread rapidly among computers on the same network, encrypt files on the infected system, empowering the attackers to demand ransom from users to decrypt the files. Similar to the WannaCry virus, Petya uses the “EternalBlue” exploit against the Windows’ Server Message Block (SMB) protocol.Recommendations
Those customers who installed Microsoft security patch MS17-010 are not at risk from Petya; however, for those customers who have yet to install this patch, we recommend the installation of this patch to help safeguard their PENTAX Medical systems listed below. More details and instructions are on Microsoft’s Security Bulletin at: https://technet.microsoft.com/en-us/library/security/ms17-010.aspx .
In addition, Microsoft has released new security patches via the June 2017 security update. This security update is meant to prevent other possible Windows’ exploits from occurring. These patches can be referenced in the Microsoft Security Advisory (MSA) 4025685 at: https://technet.microsoft.com/en-us/library/security/4025685.aspx . The PENTAX Medical systems listed below support the MSA 4025685 patches.Systems that support the Microsoft Security Advisory 4025685
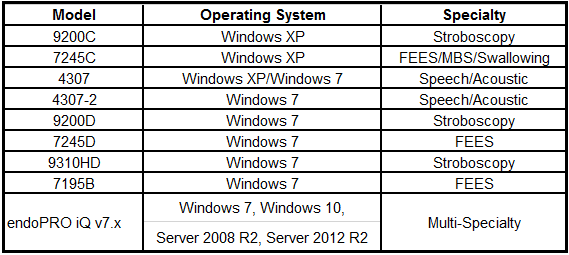
In case these products are operated in a stand-alone mode, there is no impact of the ransomware virus and there is no need to apply the security patch.
There is no impact on other products than the listed above in case they are operated in a stand-alone mode, but in case they are used in a network, we recommend to ensure the Windows PCs’ are updated.
If you have any questions, please contact your local PENTAX Medical representative.
Sincerely,
PENTAX Medical
-
URGENT MEDICAL DEVICE REMOVAL 18-009 FG-1017 Gen 2 Pentax Medical CryoBalloon™ Controller
Dear Healthcare Professional,
This letter is to inform you that PENTAX Medical is conducting a recall/corrective action for the FG-1017 C2 CryoBalloon™ Controller. Pentax Medical is performing a removal of the affected products and recommends that affected products should not be used. Please ensure that all potential users in your facility are made aware of this notification and the recommended actions.
Product Issues
PENTAX Medical has become aware of an issue which affects the FG-1017 C2 CryoBalloon™ Controller (lot numbers as listed below). It has been noted that the Controller does not detect overpressure in the balloon during the application of non-dosing puff’s of Nitrous Oxide. This can contribute to balloon over pressurization if the intended vent lumen of the catheter is significantly occluded to prevent relieving balloon pressure due to a kinked catheter condition.
Affected Product Details
Model
Description
Lot Numbers
FG-1017
C2 CryoBalloon Controller
All
Occurrence and Safety
Controller Balloon over-pressure detection. There has been one reported case of occurrence due to the use of a severely kinked catheter for ablation where the vent lumen was significantly occluded. There are currently a total of 36 Controllers remaining in distribution in the USA. If a patient is exposed to higher than physiologic pressures, adverse events such as perforation or mucosal laceration may occur.
Customer Instructions
Enclosed with this letter is a Field Action Response Form identifying the affected device(s), (lot number and quantity) which have been sold to your facility. Please forward this letter and the enclosures to the department where the above referenced items are used. The end user of the affected products should:
1. Complete, sign, and return the Field Action Response Form by either of the following methods;
- Fax to PENTAX Recall Coordinator at 201-799-4063 (alternate 201-391-4189) or
- Email a pdf copy to customeradvisories@pentaxmedical.com
2. Send a copy of the completed Response Form with the return material using the enclosed label to;
Pentax of America Inc,
303 Convention Way Suite 1
Redwood City, CA 94063
Attention: 2018-009-R ActionContact Information
If you have any questions regarding this action, please contact PENTAX Medical Customer Service.
Tel: 650-318-5899 (8:30 AM – 5:00 PM, Monday – Friday, PST)
Email: RMA@C2Therapeutics.com
PENTAX Medical regrets any inconvenience that may result from this action and appreciates your patience as we provide replacement devices. Please be assured that maintaining patient safety and quality is our utmost priority.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
Sincerely, PENTAX Medical Regulatory Affairs
-
URGENT MEDICAL DEVICE REMOVAL 18-007 Gen 2 Pentax Medical CryoBalloon™ Catheters: FG-1028 Focal Standard, FG-1024 Standard Focal Pear, and FG-1030 Standard 90
Dear Healthcare Professional,
This letter is to inform you that PENTAX Medical is conducting a recall/corrective action for Gen 2 C2 CryoBalloon™ Catheters:FG 1028 Focal Standard, FG-1024 Standard Focal Pear, and FG 1030 Standard 90 for Lot numbers as listed below. Pentax Medical is performing removal of the affected products and recommends that affected products should not be used. Please ensure that all potential users in your facility are made aware of this notification and the recommended actions.
Product Issues
PENTAX Medical has become aware of an issue which affects Gen 2 CryoBalloon™ Catheters, catalog numbers; FG-1028, FG-1024, and FG-1030 This issue is attributed to the catheter RFID tag not being correct or not being correctly or completely readable by the controller. Being that an incorrect default dose and dose increments may be transmitted to the controller it is possible that the User would not notice the inconsistency and may treat the patient using a significantly higher dose than intended.
Affected Product Details
Model
Description
Lot Numbers
FG-1028
Gen 2 CryoBalloon Catheter, Focal Standard
06152018-02, 06252018-03, 07062018-01, 07232018-02, 08102018-01, 08172018-06, 08292018-04, 9172018-01
FG-1024
Gen 2 CryoBalloon Catheter, Standard Focal Pear
06152018-01, 07172018-02, 07022018-04, 08212018-01, 08102018-03
FG-1030
Gen 2 CryoBalloon Catheter, Standard 90
08062018-03
Occurrence and Safety
RFID Tag dose values. There have been (3) reported cases to date in which the Gen 2 CryoBalloon™ Catheter FG-1028 exhibited the RFID Dose error described above. This occurred in 3 out of 239 units shipped since August 17th 2018. This is 3/239 or 1.2% of the distributed population.There are currently a total of 195 units for FG-1024, FG-1028, and FG-1030 remaining in distribution in the USA, many of which may not exhibit the issue and may already have been used, discarded or returned. If a patient is exposed to longer than desired cryoablation, adverse events such as bleeding, esophageal stricture or perforation may occur.
Customer Instructions
Enclosed with this letter is a Field Action Response Form identifying the affected device(s), (lot number and quantity) which have been sold to your facility. Please forward this letter and the enclosures to the department where the above referenced items are used. The end user of the affected products should:
1. Complete, sign, and return the Field Action Response Form by either of the following methods;
- Fax to PENTAX Recall Coordinator at 201-799-4063 (alternate 201-391-4189) or
- Email a pdf copy to customeradvisories@pentaxmedical.com
2. Send a copy of the completed Response Form with the return material using the enclosed label to;
Pentax of America Inc,
303 Convention Way Suite 1
Redwood City, CA 94063
Attention: 2018-007-R ActionContact Information
If you have any questions regarding this action, please contact PENTAX Medical Customer Service.
Tel: 650-318-5899 (8:30 AM – 5:00 PM, Monday – Friday, PST)
Email: RMA@C2Therapeutics.com
PENTAX Medical regrets any inconvenience that may result from this action and appreciates your patience as we provide replacement devices. Please be assured that maintaining patient safety and quality is our utmost priority.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
Sincerely,
PENTAX Medical Regulatory Affairs
-
URGENT MEDICAL DEVICE CORRECTION AND REMOVAL 18-001 for United States Customers Only of PENTAX Medical Duodenoscope Model ED-3490TK
Re: PENTAX Medical Duodenoscope Model ED-3490TK
Replacement of Forceps Elevator Mechanism, O-Rings, Distal End Covering, and Operation Manual UpdateDear Healthcare Professional,
This letter is to inform you that PENTAX Medical (“PENTAX”) is conducting a voluntary recall/corrective action of all ED-3490TK duodenoscopes in order to replace the forceps elevator mechanism, O-rings, and distal end covering. In January 2017, PENTAX informed ED-3490TK customers about a potential issue associated with the distal end of the ED-3490TK. The January 2017 customer letter offered recommendations intended to reduce the potential risk for contamination and subsequent patient injury and also informed customers that PENTAX personnel would be conducting a no-charge duodenoscope inspection process of the distal end covering.
The ED-3490TK duodenoscope is a flexible gastrointestinal endoscope used in procedures such as endoscopic retrograde cholangiopancreatography (ERCP). PENTAX has worked closely with the U.S. Food and Drug Administration (FDA) to mitigate the potential risk of infection in flexible endoscopy, and is conducting this field action with the knowledge of the FDA.
On February 7, 2018, FDA cleared an updated design for the ED-3490TK duodenoscope (K161222). The current voluntary recall/corrective action is being taken to replace the forceps elevator mechanism, the O-rings, and the distal end covering with materials and processes consistent with the design features of the cleared updated ED-3490TK. In addition, a periodic duodenoscope inspection process is being implemented for the forceps elevator mechanism, and is described in the updated Operation Manual (S206 R00) which is enclosed with this letter. The updated information can be found on page 22 of the Operation Manual. The Reprocessing Instructions For Use (S041 R02) have not changed and should be closely followed.
Customer Instructions:
Enclosed with this letter is a new Operation Manual (S206 R00) and a Field Correction Response Form (MK-1065 Rev B). Please discard all old copies of the ED-3490TK Operation Manual, and replace with the new Operation Manual enclosed in this package. Additional copies of the Operation Manual can be downloaded from the PENTAX Medical (USA) website located at http://www.pentaxmedical.com/pentax/en/99/1/Customer-notices. The Reprocessing IFU has not changed.
The response form identifies the serial numbers of the affected duodenoscopes which have been sold to your facility. Please complete this form, and return it to PENTAX Medical using the e-mail address or fax number listed below. Upon return of the response form, PENTAX will contact your facility to arrange the return of the ED-3490TK for the forceps elevator mechanism, O-rings and distal end covering updates. Loaner units will be supplied to customers as needed. You can continue to use your ED-3490TK duodenoscope until you are contacted to update your unit. PENTAX will continue to conduct distal tip inspections every 6 months on units that have not been updated.
PENTAX reminds its customers of the importance of using the ED-3490TK according to its current labeling. Customers must ensure that all reprocessing personnel are knowledgeable and thoroughly trained on the current Operation Manual and Reprocessing IFU for these devices. Meticulous cleaning of the elevator recesses and attention to following all reprocessing instructions are required. Additionally, PENTAX recommends that you immediately remove from use any ED-3490TK duodenoscope that shows visible signs of wear or physical damage. Continuing to use devices with integrity issues (i.e.; holes, cracks, kinks, and scratches) can contribute to persistent device contamination and subsequent patient infection.
Adverse events experienced with the use of this product should be reported as soon as possible to PENTAX at vigilance@pentaxmedical.comand the FDA’s MedWatch Adverse Event Reporting program.
Contact Information:
PENTAX regrets any inconvenience that this action may cause and appreciates your understanding and cooperation. PENTAX will issue additional communications as further information becomes available. Please be assured that maintaining patient safety and quality is our utmost priority.
If you have any questions regarding this action, please feel free to contact us at:
Tel: 1-800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST)
Fax: 201-799-4063 (alternate 201-391-4189)
Email: customeradvisories@pentaxmedical.comSincerely,
PENTAX Medical
ED-3490TK Operation Manual
ED-3490TK Elevator & O-Ring Field Action Customer Letter
ED-3490TK Field Correction Response FormMK-1062 Rev B
-
U.S. URGENT FIELD CORRECTION 17-003 Notification of Update to Instructions for Use (IFU) Ultrasound Video Gastroscope Models EG-3670URK and EG-3870UTK
May 23, 2017
Dear Valued Customer,
PENTAX Medical has become aware of an error in the Instructions for Use (IFU) for ultrasound video gastroscope models EG-3670URK and EG-3870UTK. Please ensure that all potential users in your facility are made aware of this safety notification and the recommended actions.
Affected Product Details:
Product: Ultrasound Video Gastroscope Models EG-3670URK and EG-3870UTK
Instructions for Use: Z845 Revisions 13 and 14 (R13 and R14)
Safety Instructions:
Although no incidents related to this error have been reported to PENTAX Medical, please be informed about the following error and correction:
Table 1:
Page
Error
Correct
p70
WARNING:
The cleaning detergent solution should remain in contact with the ALL internal channels and external endoscope surfaces for the time period recommended by the manufacturer of the disinfectant.
WARNING:
The disinfecting solution must remain in contact with the ALL internal channels and external endoscope surfaces for the time period recommended by the manufacturer of the disinfectant.
Customer Instructions:
In order to ensure the proper use and cleaning of the affected PENTAX endoscopes, please have users read and carefully follow the IFU addendum that is included with this letter. If a new IFU is needed, please visit PENTAX Medical’s IFU portal (https://ifu.pentaxmedical.com/welcome-to-pentax-medical/). We invite you to contact your PENTAX Medical sales representative to arrange for training regarding these new procedures.
Also enclosed with this letter is a field correction response form. The form identifies the affected endoscopes (model and serial numbers) which have been sold to your facility. Please forward this letter and the enclosures to the department in which the above referenced items are in use. The end user of the affected products should complete this form and return it to PENTAX Medical.
Contact Information
If you have any questions regarding this action, please feel free to contact PENTAX Medical Customer Service.
Tel: 800-431-5880 (8:30 AM – 5:00 PM, Monday – Friday, EST)
Fax: 201-799-4063 (alternate 201-391-4189)
Email: customeradvisories@pentaxmedical.com
This corrective action is being made with the knowledge of the U.S. Food and Drug Administration.
PENTAX Medical regrets any inconvenience that may result from this action and appreciates your patience as we introduce these updated instructions. Please be assured that maintaining patient safety and quality is our utmost priority.
Sincerely,
PENTAX of America Inc.
Director of Regulatory AffairsEnclosures:
Customer Response Form, Control Number MK-951
Ultrasound Gastroscope IFU (Z845) Addendum, MK-958 Rev. AFor a PDF copy of this Urgent Field Correction letter, please click here.
Supplementary Documents
